Open, accessible platform for microbial bioinformatics
Pre-process
Examine and improve the read quality
Analyze
Run an analysis pipeline for your data
Explore
Run additional analysis and visualizations

survey or shotgun metagenomics
viral (SARS-CoV-2)
Short read quality check
Nephele provides a pre-processing quality check pipeline for demultiplexed paired-end and single-end FASTQ files. Please see this FAQ on why you may want to run QC pipeline before you run a microbiome analysis. The Nephele QC pipeline can run a quality control check (FastQC), trim primers and/or adapters, trim and/or filter reads based on quality scores, merge read pairs, and provides summary graphs of the QC steps.
Learn more Start job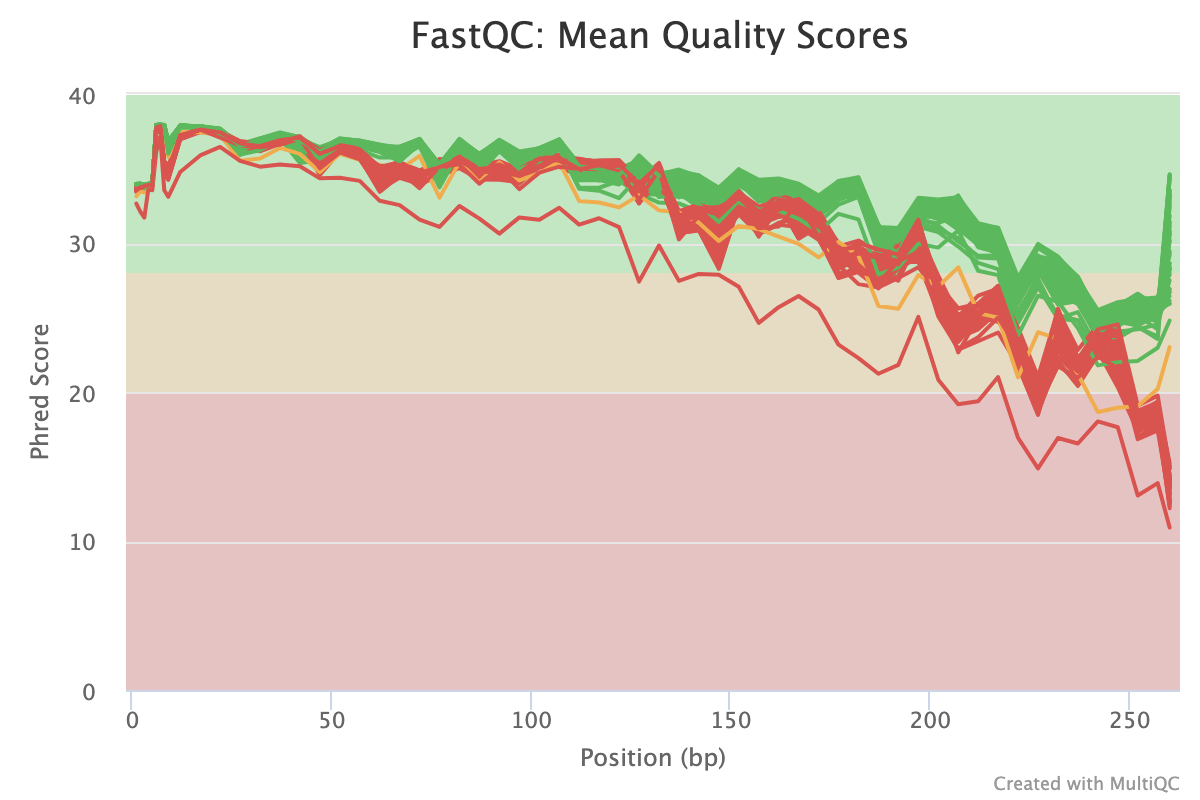
Nanopore quality check
Nephele provides a pre-processing quality check pipeline for Oxford Nanopore Technology (ONT) long read sequences. The Nephele NanoporeQC pipeline runs a quality control check (NanoPlot), trims known or unknown primers and/or adapters (Porechop_abi), trims and/or filters reads (nanoq), and provides summary graphs of the QC steps.
Learn more Start job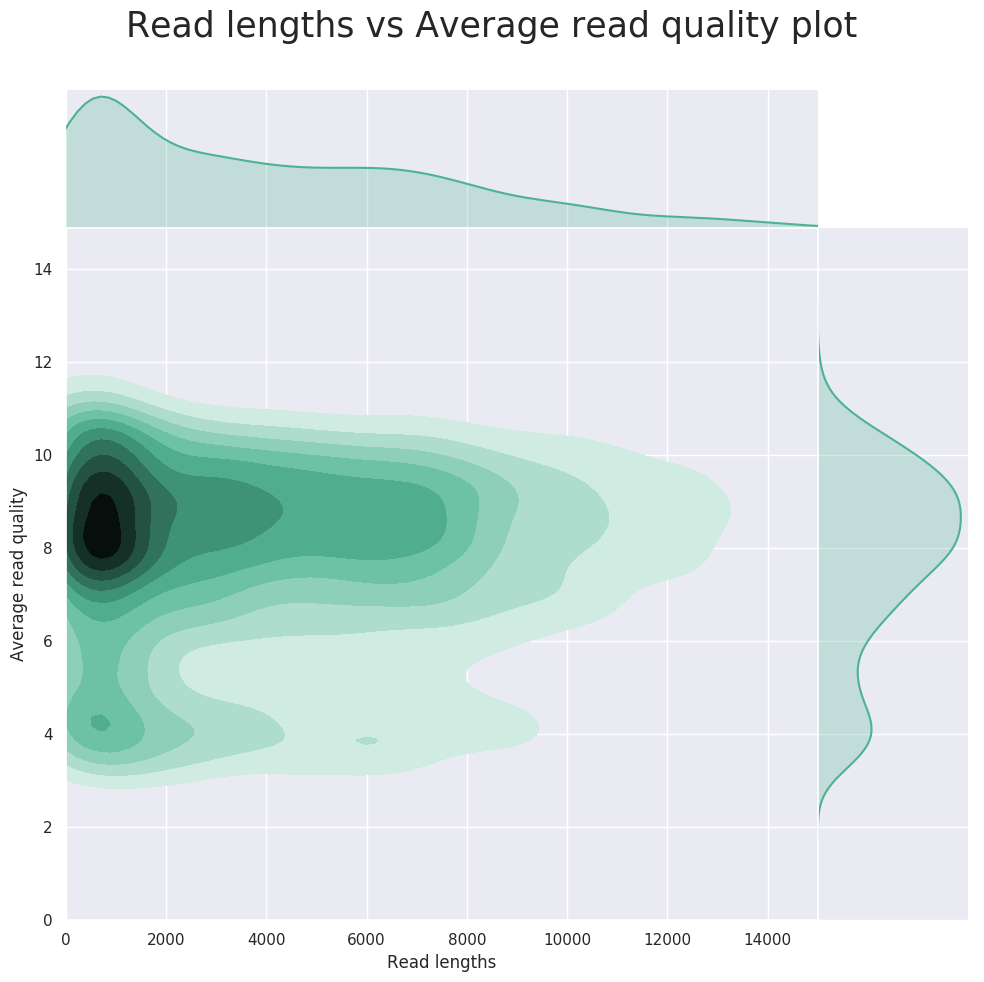
Amplicon metagenomics
Paired-end only
mothur
The mothur pipeline only operates on amplicon paired-end FASTQ data. Limited to datasets below 2GB zipped.
Learn more Start jobPaired-end or Single-end
dada2 recommended
The DADA2 pipeline accepts amplicon single or paired-end FASTQ data, and generates sequence variants with their sample-wise abundances after removing substitution and chimera errors. The pipeline provides rarefaction plots, taxonomy barplots and a biom file for use in the Downstream Analysis pipeline. Also, see Where is QIIME2?
Learn more Start jobDADA2 ITS
The DADA2 ITS pipeline accepts ITS amplicon FASTQ data, and generates sequence variants with their sample-wise abundances after removing substitution and chimera errors. The pipeline provides rarefaction plots, taxonomy barplots and a biom file for use in the Downstream Analysis pipeline.
Learn more Start jobqiime2/vsearch
The QIIME2/VSEARCH 16S pipeline starts with FASTQ data and finishes with a rarefaction plot, taxonomy barplot and a biom file that can be further explored using the Downstream Analysis pipeline. It relies on VSEARCH for De novo, Open, and Closed reference clustering into OTUs. For Single-End data, it also provides the DEBLUR denoising algorithm.
Learn more Start jobShotgun metagenomics
Paired-end only
WGSA2 recommended
The WGSA2 pipeline uses paired-end shotgun reads to produce de novo assemblies for each sample in a metagenomic dataset and extract taxonomic and functional information about the microbial community.
Learn more Start jobPaired-end or Single-end
WGS - bioBakery
The pipeline runs the BioBakery Whole Metagenome Shotgun (wmgx) for taxonomic profiling and functional annotation, and Visualization for Whole Metagenome Shotgun (wmgx_vis) bioBakery workflows. It is assembly-free.
Learn more Start jobSingle-end
LoRA
The long read assembly-based pipeline uses ONT or PacBio long sequence reads from metagenomic datasets, to produce longer de novo assemblies per sample, taxonomic and functional profiles and other features of the microbial community.
Learn more Start jobViral genomics
Paired-end or Single-end
sars-cov-2
This pipeline assembles SARS-CoV-2 genome and calls mutations from Illumina sequence data generated using a tiled multiplexed primers strategy (example: Artic protocol). Users can select from primers such as ARTIC and NEB-Varskip. Alternatively, users can upload a custom primers design (in .bed format). See more info below.
Learn more Start jobDownstream Analysis: Diversity
You can use biom files from your 16S or ITS pipeline outputs to run the downstream analysis (DA) pipeline. Nephele's DA pipeline uses QIIME 2 to provide sample observation and taxonomic summaries and diversity analyses of an OTU table.
Learn more Start job
Viral metagenomics: DiscoVir
The DiscoVir pipeline uses metagenome assemblies (.fasta files) and sequence alignments (.bam files) to identify viral sequences, taxonomically and functionally characterize the virome, and predict phage hosts.
Learn more Start job
Metagenome Inference: PICRUSt2
This pipeline uses PICRUSt2 to predict the functional potential of a community based on marker gene sequencing profiles. It is compatible with the outputs from OTU-picking or denoising algorithm employed in QIIME2 and DADA2 pipelines. The output folder contains inferred genes and protein families and tables of predicted KEGG orthologs (KO), Enzymes (EC) and pathways.
Learn more Start job
Or
Search and View your job history in My Workspace