QIIME 2 Pipeline
The QIIME 2 pipeline is intended to be an upgrade of the QIIME 1 (v1.9) pipeline. Back in 2016, Greg Caporaso and the QIIME team started announcing their new design for QIIME and the intentions to transition and expand functionality through plugins in the QIIME 2. Learn more here. The pipeline's steps are below, and see the output files for a description of the results and how you can view and explore them on your own.
Pipeline steps
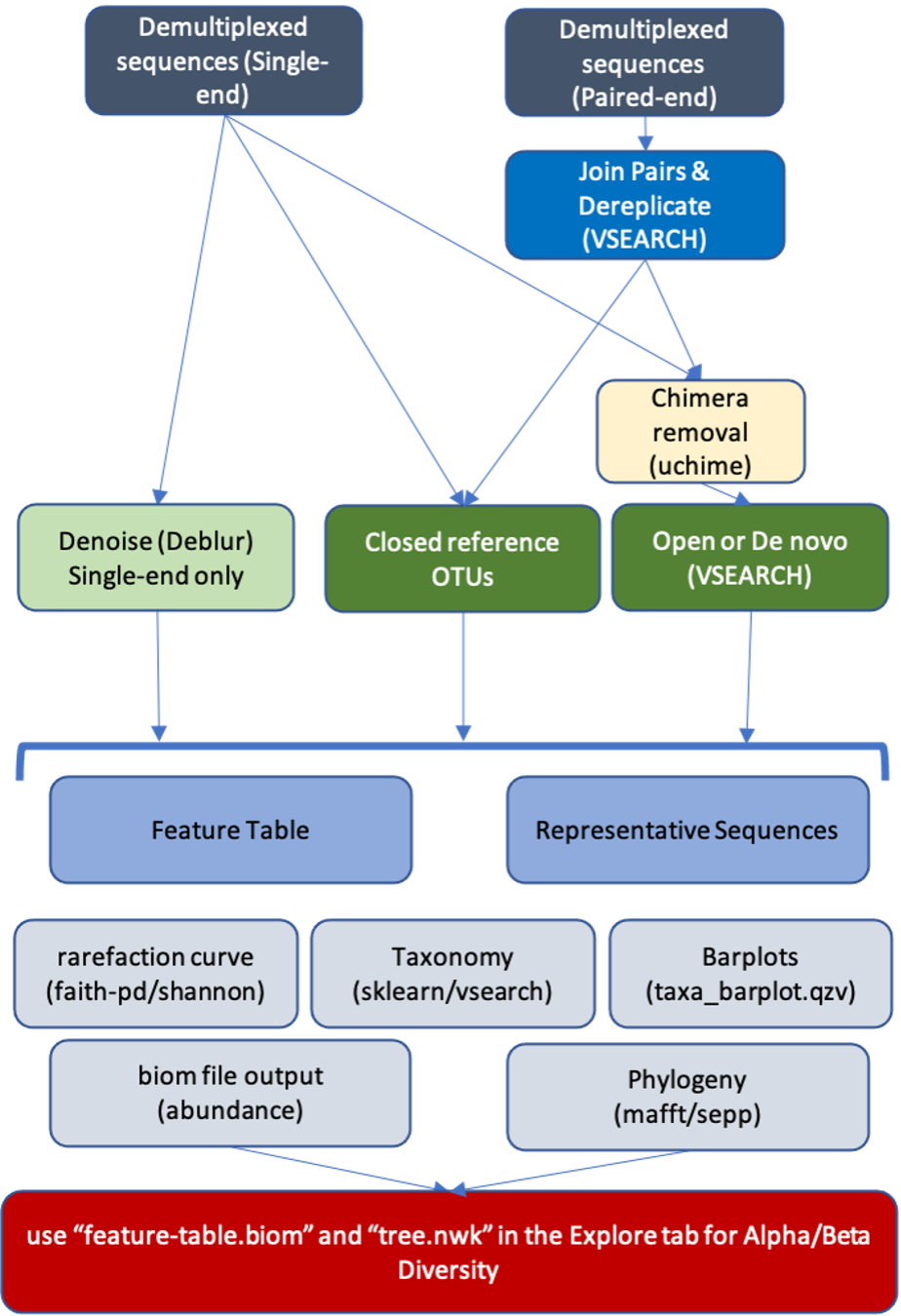
We generally follow the workflows recommended by the QIIME 2 developers described here, but please see details and diagram below for the specific steps that we have chosen to include.
Nephele’s QIIME 2 pipeline takes single or paired-end FASTQ files as input. Ideally, you would have first verified the quality of the sequence files (Hint: use the Pre-process tab). In this pipeline, the paired-end reads get merged, filtered by quality and then dereplicated using VSEARCH. Similarly, the single-end reads first get trimmed, quality filtered, and dereplicated.
In the next step, if you have single-end data, you will need to choose between the clustering method (VSEARCH) or the denoising method (Deblur) for error correction (see green boxes in diagram below). Paired-end data will automatically be processed using VSEARCH's clustering method. Nephele’s QIIME 2 pipeline offers you the new OTU clustering options available via the QIIME 2 plugins, which correspond to the Open Reference, Closed Reference and De Novo clustering methods used heavily in QIIME 1 pipeline. The clustering and denoising methods generate a table of features with frequency and sequences.
The pipeline will then align OTU sequences and generate a phylogenetic tree, and create a rarefaction curve labeled based on the metadata columns provided.
-
In the final steps of the pipeline, the sequences will be classified by your choice of method (sklearn or VSEARCH) and database (SILVA or Greengenes), and a barplot of taxa relative abundances will be generated. See the output files section for a description of the results and how you can view and explore them on your own.
User inputs
- FASTQ files of raw sequence reads after demultiplexing. The pipeline accepts single-end and paired-end data. See these FAQs for more help: How do I know if my data is paired-end or single-end? and How do I know if my data is demultiplexed?
- Mapping file describing sample names and metadata.
User options
Parameters for single-end reads only
-
Sequence Error Correction: Choose which method to use: Denoising using Deblur or Clustering using VSEARCH.
Important note: Deblur's method is only for single-end data. For denoising paired-end data, consider Nephele's DADA2 pipeline which uses the dada algorithm for denoising. Even though the QIIME 2 developers also offer a DADA2 plugin, the Nephele team has decided to have a separate DADA2 pipeline written using the native R package directly.
-
Trim Length (for Denoising only):
If desired, trim the length of the single read or disable trimming by entering -1
Note: Deblur only works with sequences that are the same length therefore please ensure that the dataset doesn't contain sequences shorter than the trim length you choose
Parameters for Paired-end and Single-end reads
- Minimum Phred Quality Score: Use this score to filter reads of low quality
- Clustering algorithm: Select from Open, Closed, Denovo OTU clustering. For paired-end data or if VSEARCH clustering is chosen as the error correction method for single-end reads.
- Chimeras Removal: check the box to run uchime for chimera detection and removal
-
OTU reference: Selection of database of representative sequences from OTUs generated through clustering of Greengenes
sequences (release 13_8) at 85, 97 and 99% identity. Use 85% identity only for a quick clustering inspection.
Best to use 97 or 99%
-
Phylogenetic Tree: Choose mafft to align sequences and infer a phylogenetic tree using FastTree. Choose sepp to insert fragment sequences into
reference phylogenies using databases previously generated from SILVA v128 or Greengenes (13_8)
-
Taxonomy classification method: Choose VSEARCH the perform VSEARCH global alignment between query and reference_reads from SILVA
database (v138), then assign consensus taxonomy to each query sequence. Choose sklearn to classify reads using machine learning
and pre-trained classifier
- sklearn options: Choose database from among the V4 region or full length of Greengenes v13_8 or SILVA v138 trained classifiers
- Barplots Filter Samples Minimum Frequency
- Perc Identity (for Clustering only): Percent identity threshold for constructing OTUs
Output Files/Directories
The output files will include several ".qzv" files that can be visualized by "drag and drop" on the QIIME 2 View page and plain text/tabular files (.txt). The pipeline also generates a BIOM file and ".nwk" tree files that you could bring into the Explore section of Nephele to be the input for the Downstream Analysis pipeline where you will be able to run alpha/beta diversity and other analyses.
- logfile.txt: contains the messages associated with the pipeline
- logfile_debug.txt: expanded file useful for debugging
- taxa_barplot.qzv: barplot of relative abundance of taxa
- taxonomy.qzv: visual representation of table of taxonomy association for each feature (OTU)
- feature-table.biom: frequency table (abundance for each OTU) in BIOM V2 format. Can be used as input with the Downstream Analysis pipeline.
- feature-table.tsv: counts and taxonomic assignment of OTUs in each sample.
- taxonomy.tsv: text file of taxonomy association
- alpha-rarefaction.qzv: visual representation of rarefaction curve based on alpha diversity metrics
- rep-seqs.qzv: visual representation of representative sequences for each feature identified
- table.qzv: visual representation of the biom file
- tree.nwk: tree file in newick format. The folders named rooted-tree and unrooted-tree, each contain the respective tree file
- dna-sequences.fasta: this file contains the sequences of the representative sequences resulting from the VSEARCH clustering or Deblur denoising steps used subsequently in the alignment step for phylogeny analysis
- Other files ending on .qza: QIIME 2 artifacts which contains data and metadata. These automatically track the type, format, and provenance of data for researchers.
Dependencies
- System versions: QIIME 2 release: 2022.2
-
Required plugins
- alignment: 2022.2
- deblur: 2022.2
- demux: 2022.2
- diversity: 2022.2
- feature-classifier: 2022.2
- feature-table: 2022.2
- fragment-insertion: 2022.2
- metadata: 2022.2
- phylogeny: 2022.2
- quality-control: 2022.2
- quality-filter: 2022.2
- sample-classifier: 2022.2
- taxa: 2022.2
- vsearch: 2022.2
References
In order to cite QIIME 2 and plugins properly, please follow recommendations from QIIME 2 developers here. One easy way to get all relevant references is to use the taxonomy.qzv file and view "Citations" when you view the Provenance of the file in the QIIME 2 View page.
- QIIME 2: Bolyen, Evan, Jai Ram Rideout, Matthew R. Dillon, Nicholas A. Bokulich, Christian C. Abnet, Gabriel A. Al-Ghalith, Harriet Alexander, et al. "Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2." Nature Biotechnology, July 24, 2019. https://doi.org/10.1038/s41587-019-0209-9.
-
QUALITY FILTERING: Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, J. I., Knight, R., Mills, D. A., & Caporaso, J. G. (2013). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nature methods, 10(1), 57–59. https://doi.org/10.1038/nmeth.2276.
-
VSEARCH: Rognes, Torbjørn, Tomáš Flouri, Ben Nichols, Christopher Quince, and Frédéric Mahé. "VSEARCH: A Versatile Open Source Tool for Metagenomics." PeerJ 4 (October 18, 2016): e2584. https://doi.org/10.7717/peerj.2584.
-
UCHIME (method used by VSEARCH for chimera removal): Robert C. Edgar, Brian J. Haas, Jose C. Clemente, Christopher Quince, Rob Knight, UCHIME improves sensitivity and speed of chimera detection, Bioinformatics, Volume 27, Issue 16, 15 August 2011, Pages 2194–2200, https://doi.org/10.1093/bioinformatics/btr381.
-
SKLEARN: Bokulich, N.A., Kaehler, B.D., Rideout, J.R. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90 (2018). https://doi.org/10.1186/s40168-018-0470-z.
-
DEBLUR: Amnon Amir, Daniel McDonald, Jose A Navas-Molina, Evguenia Kopylova, James T Morton, Zhenjiang Zech Xu, Eric P Kightley, Luke R Thompson, Embriette R Hyde, Antonio Gonzalez, and Rob Knight. Deblur rapidly resolves single-nucleotide community sequence patterns. MSystems, 2(2):e00191–16, 2017.
-
MAFFT: Katoh, Kazutaka, and Daron M Standley. "MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability." Molecular Biology and Evolution 30, no. 4 (2013): 772–80. https://doi.org/10.1093/molbev/mst010.
-
FASTTREE: Price, Morgan N, Paramvir S Dehal, and Adam P Arkin. "FastTree 2–Approximately Maximum-Likelihood Trees for Large Alignments." PloS One 5, no. 3 (2010): e9490. https://doi.org/10.1371/journal.pone.0009490.
-
SEPP: Stefan Janssen, Daniel McDonald, Antonio Gonzalez, Jose A. Navas-Molina, Lingjing Jiang, Zhenjiang Zech Xu, Kevin Winker, Deborah M. Kado, Eric Orwoll, Mark Manary, Siavash Mirarab, and Rob Knight. Phylogenetic placement of exact amplicon sequences improves associations with clinical information. mSystems, 2018. doi:10.1128/mSystems.00021-18.